





Abstract
Systems biotechnology has been established as a highly potent tool for bioprocess development in recent years. The applicability to complex metabolic processes such as protein synthesis and secretion, however, is still in its infancy. While yeasts are frequently applied for heterologous protein production, more progress in this field has been achieved for bacterial and mammalian cell culture systems than for yeasts. A critical comparison between different protein production systems, as provided in this review, can aid in assessing the potentials and pitfalls of applying systems biotechnology concepts to heterologous protein producing yeasts. Apart from modelling, the methodological basis of systems biology strongly relies on postgenomic methods. However, this methodology is rapidly moving so that more global data with much higher sensitivity will be achieved in near future. The development of next generation sequencing technology enables an unexpected revival of genomic approaches, providing new potential for evolutionary engineering and inverse metabolic engineering.
Introduction
The unexpectedly fast progress in genome sequencing over the last decade has provided an invaluable source of information on the physiology of microorganisms, including a comprehensive overview on cellular endowment with metabolic enzymes. Simultaneously, metabolic modelling has been developed and applied to the mathematical description of the central metabolic processes of bacteria (Edwards & Palsson, 2000) and yeast (Förster, 2003). Together with extensive work on genomic data to address ideally all metabolic processes of a cell, these metabolic models led to the concept of systems biology (Westerhoff & Palsson, 2004). Several systems biology models of Saccharomyces cerevisiae have previously been described and recently unified to one comprehensive model (Herrgård, 2008, and references therein). To acquire data for dynamic modelling, postgenomic analyses at the transcriptomics, proteomics and metabolomics level are implemented.
These models offer the opportunity to predict cellular processes and are therefore regarded as highly valuable resources for strain optimization. Since 1990, the concepts of metabolic engineering have been developed and applied as the knowledge-based improvement of cell factories using genetic engineering (Bailey, 1991; Nielsen, 2001). Extending the concepts of metabolic engineering to a broad system basis has led to the conception of systems biotechnology (Lee, 2005; Nielsen & Jewett, 2008). While systems biology aims ideally at the global understanding and modelling of the entire cellular network of reactions, systems biotechnology will rather accept gaps in the description of cellular processes, as long as the processes related to product formation can be mapped. The systems biotechnology approach can be seen as an iterative, cyclic process, integrating high throughput data generation with metabolic modelling and production strain optimization (Fig. 1).
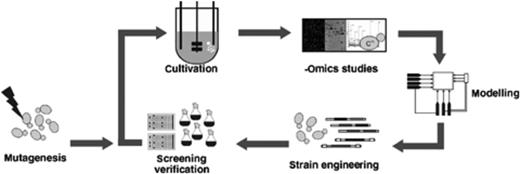
The systems biotechnology circle. A primary production strain is cultivated under relevant conditions. Omics methods feed models, which aid the design of strain engineering and screening of new, improved strains. Random mutagenesis serves to increase variability, which can also be achieved by evolutionary engineering.
The concepts of metabolic engineering were initially mostly applied to the production of metabolites. In 2000, heterologous proteins were introduced as a new class of products to be addressed by metabolic engineering (Ostergaard, 2000). It is obvious that the complexity of the protein production and secretion process (Fig. 2) renders it much more challenging to be addressed by tools of rational and quantitative analysis, as summarized in Table 1.
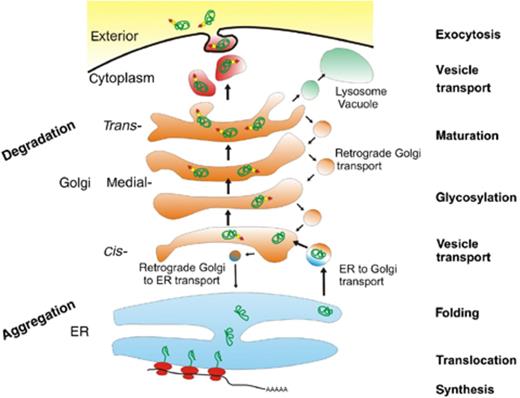
Protein synthesis and secretion in eukaryotic cells. Pathways and key ‘metabolic’ processes of secreted proteins are indicated, including unproductive steps such as degradation or aggregation. These pathways should serve as a basis for the development of a model of the protein production and secretion process.
Challenges for systems biotechnology research for heterologous protein production
| Key elements for systems biotechnology | Challenges faced in protein expression and secretion |
| Metabolites and enzymes | Molecular players only partly defined |
| Metabolic pathways | Pathways less clear than those for metabolic processes |
| Stoichiometry of metabolic reactions | Stoichiometry difficult to define |
| Metabolic fluxes | Fluxes and concentrations of participating ‘metabolites’ difficult or not yet measurable |
| Key elements for systems biotechnology | Challenges faced in protein expression and secretion |
| Metabolites and enzymes | Molecular players only partly defined |
| Metabolic pathways | Pathways less clear than those for metabolic processes |
| Stoichiometry of metabolic reactions | Stoichiometry difficult to define |
| Metabolic fluxes | Fluxes and concentrations of participating ‘metabolites’ difficult or not yet measurable |
Challenges for systems biotechnology research for heterologous protein production
| Key elements for systems biotechnology | Challenges faced in protein expression and secretion |
| Metabolites and enzymes | Molecular players only partly defined |
| Metabolic pathways | Pathways less clear than those for metabolic processes |
| Stoichiometry of metabolic reactions | Stoichiometry difficult to define |
| Metabolic fluxes | Fluxes and concentrations of participating ‘metabolites’ difficult or not yet measurable |
| Key elements for systems biotechnology | Challenges faced in protein expression and secretion |
| Metabolites and enzymes | Molecular players only partly defined |
| Metabolic pathways | Pathways less clear than those for metabolic processes |
| Stoichiometry of metabolic reactions | Stoichiometry difficult to define |
| Metabolic fluxes | Fluxes and concentrations of participating ‘metabolites’ difficult or not yet measurable |
In cases where the genetic traits controlling complex cellular responses are not known, researchers have applied random mutagenesis and selection schemes to engineer metabolic pathways by modifying enzymes, transporters or regulatory proteins. This method has been termed evolutionary engineering (Sonderegger & Sauer, 2003), but its application to protein production is not straight forward as protein overexpression is usually not advantageous for the cell. Single cell sorting of large cell populations may be applied to overcome this limitation (Mattanovich & Borth, 2006). However, as the changes are not directed, it is often difficult to determine the genetic modification that is responsible for the improvement (Nevoigt, 2008). Understanding the biological system as a whole greatly facilitates the rational understanding of such mutants. Genome-wide analysis methods also made another biotechnological approach, namely inverse metabolic engineering (Bailey, 1996), much more feasible, where phenotypic differences serve as the basis for elucidating genetic modifications needed to optimize production strains (Bro & Nielsen, 2004).
Yeasts are attractive hosts for production of heterologous proteins (Porro, 2005). However, a number of bottlenecks and stress factors limit the full potential of this class of organisms (Mattanovich, 2004), and systems biotechnology will offer new opportunities for modelling, analysis and optimization of protein production systems. In the following we will describe the applications of systems biotechnology to yeasts with a strong emphasis on heterologous protein production, highlighting work on other classes of host organisms also, and provide an outlook as to where the development and integration of new methodology can lead this field.
Impact of systems biology on yeast biotechnological processes
Systems biology is not a purely academic research area as the quantitative description of microbial production cell lines is also already of interest for biotechnological industry. As pointed out before, yeasts producing heterologous proteins have rarely been investigated on a systems level so far, while the importance of systems biotechnology for industrial applications others than protein production has been well documented in the recent years (Pizarro, 2007; Takors, 2007; Mukhopadhyay, 2008). These applications span mainly the production of primary metabolites such as amino acids, alcohols or organic acids, often employing bacteria as production hosts. Global analysis of the host cell metabolism can help to improve the production of metabolites, which may consequently require further cell engineering to resist high concentrations of possibly toxic chemical compounds. Alper (2006) recently showed how engineering of the global transcription machinery can help to improve ethanol resistance in yeast cells.
Additionally, systems biotechnology already impacts on the production of yeast-based alcoholic beverages as a system-wide understanding of the molecular basis of the production process can lead to improved sensory qualities for consumer demands. Proteomic and transcriptomic methods have been applied to investigate wine and beer fermentations (Kobi, 2004; Beltran, 2006; Zuzuarregui, 2006). It becomes clear from the system-wide analysis of wine fermentations that microbial cells encounter many different kinds of stresses during the fermentation process. However, it is obvious that a deeper understanding of the cellular reactions to environmental stresses is also crucial for other biotechnologically relevant batch and fed-batch processes such as amino acid, biofuel and, of course, protein production. The yeast stress response has been a topic of detailed investigations in the recent years. Both, transcriptomic and proteomic approaches have been used to investigate the effect of temperature (Gasch, 2000; Tai, 2007), high osmolarity (Blomberg, 1995; Chen, 2003; Gori, 2007; Kim, 2007), hydrostatic pressure (Fernandes, 2004) and nutrient limitations (Kolkman, 2006) in several yeast and fungal species in recent years. It has been outlined earlier that cellular reactions to environmental stresses are mainly a transient response, on which most studies have focused. However, during industrial processes, rather constant suboptimal growth conditions that are far from the natural environment of the cells are imposed (Mattanovich, 2004). For example, several reports indicate a positive effect of reduced growth temperature on the production rate of heterologous proteins in the yeast Pichia pastoris (Li, 2001; Jahic, 2003; Shi, 2003). However, these data are not fully conclusive as the authors suggest that lower activity of proteases in the culture supernatant or decreased cell death rate is responsible for increases in productivity. On the other hand, Hohenblum (2003) showed that, at least for P. pastoris, significant cell death occurs at low pH while temperature does not influence viability. None of these studies applied system-wide analysis of the host organism, so that the underlying biology remained unexplored. Newer studies may shed light on such contradictory data. Recently, Tai (2007) performed steady-state cultivations of S. cerevisiae at different temperatures and analysed them with microarrays. Although not using heterologous protein-producing strains, these experiments allow conclusions on the long-term adaptation of production cells to suboptimal conditions. The authors observed an upregulation of ribosome biogenesis genes and a downregulation of environmental stress response genes at a low temperature, which differed largely from previous results on rapid changes of temperature. Similarly, Gasser (2007a) found stress response genes downregulated at lower temperature in steady-state cultures of P. pastoris expressing an antibody Fab fragment, while the specific productivity of this protein was increased.
However, transcriptome, proteome and metabolome data from small-scale laboratory experiments might differ significantly from cellular regulatory patterns as they occur during large-scale industrial processes. Furthermore there might be crucial genotypic differences between laboratory and industrial strains. As the majority of the mentioned studies were performed in laboratory strains, the direct applicability of these results on industrial strains is questionable. Production strain optimization can be achieved if the cellular metabolism and the regulatory networks of an industrial cell line are considered and investigated (Takors, 2007). Up to now, such results are rarely obtained in academia, as the genomic information of important industrial strains is still missing. Omics approaches as well as fast modern sequencing techniques bear the potential to change the methodical approaches here, as will be discussed below.
The external stresses mentioned above, and intrinsic stress mediated by protein overproduction, play a major role for the physiological constraints of a protein production system (Mattanovich, 2004). As these constraints share similar patterns among different classes of host organisms, we will also highlight research with non-yeast hosts for protein production in the following chapter.
Application of systems biotechnology to heterologous protein production
Engineering of recombinant yeasts based on genome-wide analysis
Applications of genome-wide technologies in yeasts are scarce in the field of recombinant protein production. Some of the rare examples analysing cellular responses due to protein overproduction are reported for the nonconventional yeasts P. pastoris (Sauer, 2004; Gasser, 2007a; Dragosits, 2009) and Kluyveromyces lactis (van Ooyen, 2006). The analysis of the cellular proteome during a fermentation of a chymosin expressing K. lactis strain indicated stress during protein production (upregulation of Hsp26p and Sod2p; van Ooyen, 2006), whereas the P. pastoris work outlines how environmental factors such as temperature and pH affect protein expression and secretion on a transcriptomic (Sauer, 2004; Gasser, 2007a) and proteomic level (Dragosits, 2009). Alternatively, metabolic flux analyses of protein-producing yeasts were performed. These focused, on the one hand, on the synthesis of high levels of intracellular human superoxide dismutase in S. cerevisiae (Gonzalez, 2003), and, on the other, on core metabolic processes of P. pastoris during growth on glycerol and methanol (Solà, 2004, 2007). However, apart from one exception, no strain improvement strategies resulted out of all these studies so far (Table 2).
Overview of systems biotechnological approaches for improved recombinant protein production in different hosts
| Host | Analyses | Engineering | Results | References |
| E. coli | Transcriptome, proteome | Ribosomal genes, amino acid biosynthesis genes, IbpAB | Up to fourfold higher productivity | Reviewed in Park (2005) |
| E. coli | Proteome | Controlled coexpression of PspA | Aldor (2005) | |
| E. coli | Proteome, transcriptome | Use of rare codons | Eight times more recombinant protein | Lee & Lee (2005) |
| E. coli | Proteome | New promoter (aldA) | 30-fold higher product levels | Han (2008) |
| E. coli | Secreted proteome | OsmY as fusion partner | High-level secretion | Qian (2008) |
| B. megaterium | Metabolic fluxes | Pyruvate as carbon source | 17-fold more secreted product, less protease activity | Fürch (2007) |
| S. cerevisiae | Random mutagenesis | Co-chaperone genes related to Kar2 activity | 1.5-fold increase of expression | Payne (2008) |
| P. pastoris | Transcriptome | overexpression of secretion factors | 2.5-fold increase of secretion | Gasser (2007b) |
| A. niger | ‘Genomic methods’ genome sequence and transcriptome | Disruption of protease genes | 1.4-fold increased secretion | Wang (2008) |
| A. niger | Transcriptome and proteome ‘integrative genomics’ | Knock-out of ERAD factor doaA and overexpression of oligosaccharyltransferase sstC | Improved intracellular production | Jacobs (2009) |
| A. oryzae | Transcriptome | Knock-down of protease genes | 1.2-fold increased secretion | Kimura (2008) |
| CHO | Transcriptome | Overexpression of antiapoptotic and knock-down of proapoptotic genes | Higher viability leading to 2.5-fold higher titers | Wong (2006) |
| CHO | Transcriptome | Stress markers for early clone screening | Time for clone establishment? Better and earlier clone selection | Trummer (2008) |
| NS0 | Transcriptome, proteome | Genes related to cholesterol dependence | Cholesterol-independent cell lines | Seth (2006) |
| Host | Analyses | Engineering | Results | References |
| E. coli | Transcriptome, proteome | Ribosomal genes, amino acid biosynthesis genes, IbpAB | Up to fourfold higher productivity | Reviewed in Park (2005) |
| E. coli | Proteome | Controlled coexpression of PspA | Aldor (2005) | |
| E. coli | Proteome, transcriptome | Use of rare codons | Eight times more recombinant protein | Lee & Lee (2005) |
| E. coli | Proteome | New promoter (aldA) | 30-fold higher product levels | Han (2008) |
| E. coli | Secreted proteome | OsmY as fusion partner | High-level secretion | Qian (2008) |
| B. megaterium | Metabolic fluxes | Pyruvate as carbon source | 17-fold more secreted product, less protease activity | Fürch (2007) |
| S. cerevisiae | Random mutagenesis | Co-chaperone genes related to Kar2 activity | 1.5-fold increase of expression | Payne (2008) |
| P. pastoris | Transcriptome | overexpression of secretion factors | 2.5-fold increase of secretion | Gasser (2007b) |
| A. niger | ‘Genomic methods’ genome sequence and transcriptome | Disruption of protease genes | 1.4-fold increased secretion | Wang (2008) |
| A. niger | Transcriptome and proteome ‘integrative genomics’ | Knock-out of ERAD factor doaA and overexpression of oligosaccharyltransferase sstC | Improved intracellular production | Jacobs (2009) |
| A. oryzae | Transcriptome | Knock-down of protease genes | 1.2-fold increased secretion | Kimura (2008) |
| CHO | Transcriptome | Overexpression of antiapoptotic and knock-down of proapoptotic genes | Higher viability leading to 2.5-fold higher titers | Wong (2006) |
| CHO | Transcriptome | Stress markers for early clone screening | Time for clone establishment? Better and earlier clone selection | Trummer (2008) |
| NS0 | Transcriptome, proteome | Genes related to cholesterol dependence | Cholesterol-independent cell lines | Seth (2006) |
Overview of systems biotechnological approaches for improved recombinant protein production in different hosts
| Host | Analyses | Engineering | Results | References |
| E. coli | Transcriptome, proteome | Ribosomal genes, amino acid biosynthesis genes, IbpAB | Up to fourfold higher productivity | Reviewed in Park (2005) |
| E. coli | Proteome | Controlled coexpression of PspA | Aldor (2005) | |
| E. coli | Proteome, transcriptome | Use of rare codons | Eight times more recombinant protein | Lee & Lee (2005) |
| E. coli | Proteome | New promoter (aldA) | 30-fold higher product levels | Han (2008) |
| E. coli | Secreted proteome | OsmY as fusion partner | High-level secretion | Qian (2008) |
| B. megaterium | Metabolic fluxes | Pyruvate as carbon source | 17-fold more secreted product, less protease activity | Fürch (2007) |
| S. cerevisiae | Random mutagenesis | Co-chaperone genes related to Kar2 activity | 1.5-fold increase of expression | Payne (2008) |
| P. pastoris | Transcriptome | overexpression of secretion factors | 2.5-fold increase of secretion | Gasser (2007b) |
| A. niger | ‘Genomic methods’ genome sequence and transcriptome | Disruption of protease genes | 1.4-fold increased secretion | Wang (2008) |
| A. niger | Transcriptome and proteome ‘integrative genomics’ | Knock-out of ERAD factor doaA and overexpression of oligosaccharyltransferase sstC | Improved intracellular production | Jacobs (2009) |
| A. oryzae | Transcriptome | Knock-down of protease genes | 1.2-fold increased secretion | Kimura (2008) |
| CHO | Transcriptome | Overexpression of antiapoptotic and knock-down of proapoptotic genes | Higher viability leading to 2.5-fold higher titers | Wong (2006) |
| CHO | Transcriptome | Stress markers for early clone screening | Time for clone establishment? Better and earlier clone selection | Trummer (2008) |
| NS0 | Transcriptome, proteome | Genes related to cholesterol dependence | Cholesterol-independent cell lines | Seth (2006) |
| Host | Analyses | Engineering | Results | References |
| E. coli | Transcriptome, proteome | Ribosomal genes, amino acid biosynthesis genes, IbpAB | Up to fourfold higher productivity | Reviewed in Park (2005) |
| E. coli | Proteome | Controlled coexpression of PspA | Aldor (2005) | |
| E. coli | Proteome, transcriptome | Use of rare codons | Eight times more recombinant protein | Lee & Lee (2005) |
| E. coli | Proteome | New promoter (aldA) | 30-fold higher product levels | Han (2008) |
| E. coli | Secreted proteome | OsmY as fusion partner | High-level secretion | Qian (2008) |
| B. megaterium | Metabolic fluxes | Pyruvate as carbon source | 17-fold more secreted product, less protease activity | Fürch (2007) |
| S. cerevisiae | Random mutagenesis | Co-chaperone genes related to Kar2 activity | 1.5-fold increase of expression | Payne (2008) |
| P. pastoris | Transcriptome | overexpression of secretion factors | 2.5-fold increase of secretion | Gasser (2007b) |
| A. niger | ‘Genomic methods’ genome sequence and transcriptome | Disruption of protease genes | 1.4-fold increased secretion | Wang (2008) |
| A. niger | Transcriptome and proteome ‘integrative genomics’ | Knock-out of ERAD factor doaA and overexpression of oligosaccharyltransferase sstC | Improved intracellular production | Jacobs (2009) |
| A. oryzae | Transcriptome | Knock-down of protease genes | 1.2-fold increased secretion | Kimura (2008) |
| CHO | Transcriptome | Overexpression of antiapoptotic and knock-down of proapoptotic genes | Higher viability leading to 2.5-fold higher titers | Wong (2006) |
| CHO | Transcriptome | Stress markers for early clone screening | Time for clone establishment? Better and earlier clone selection | Trummer (2008) |
| NS0 | Transcriptome, proteome | Genes related to cholesterol dependence | Cholesterol-independent cell lines | Seth (2006) |
The comparison of the differential transcriptome of a P. pastoris strain overexpressing human trypsinogen vs. a nonexpressing strain did reveal a network of genes being influenced due to the exploitation of the cellular expression machinery. This knowledge was further exploited to elucidate novel secretion helper factors that allowed the removal of bottlenecks in protein expression. Thirteen out of the 524 significantly regulated genes were selected and overexpressed in a P. pastoris strain producing a human antibody Fab fragment. Five previously characterized secretion helpers (Pdi1, Ero1, Sso2, Kar2/BiP and Hac1), as well as six novel, hitherto unidentified, factors, more precisely Bfr2 and Bmh2 involved in protein transport, the chaperones Ssa4 and Sse1, the vacuolar ATPase subunit Cup5 and Kin2, a protein kinase connected to exocytosis, increasing both specific production rates as well as volumetric productivity of an antibody fragment up to 2.5-fold in fed batch fermentations (Gasser, 2007b). Very recently, a similar approach was leading to improved membrane protein production in S. cerevisiae, based on engineered expression of BMS1, involved in ribosomal subunit assembly (Bonander, 2009). A convincing example of evolutionary engineering was based on random mutagenesis and screening for overproduction of human serum albumin in S. cerevisiae, followed by the identification of four genes related to Kar2 ATPase activity, which were upregulated in the selected mutant strain. Overexpression of these genes in other S. cerevisiae strains led to increased production of three different heterologous proteins (Payne, 2008).
Protein folding and secretion appear to be major limitations for yeast expression systems, while the main concerns for other systems are growth, viability and metabolic burden (see Mammalian cells and Bacteria). For yeast production hosts these problems are not as crucial for the production of recombinant proteins, which can be regarded as one reason why systems biotechnology-based strain engineering has hardly been applied so far in this area.
Another important aspect that explains the lack of omics-based cell engineering in yeasts is the degree of availability of omics tools for yeast and other fungal species. Out of the 82 presently sequenced ascomycetes genomes, only 15% are biotechnologically used organisms, whereas the majority (54%) were pathogens, and the remaining were sequenced for comparative genomic studies (Saccharomyces sensu stricto group). The lack of published genome sequences is reflected in a lack of commercially available microarrays for most yeasts species. Some exceptions are arrays available for S. cerevisiae and Schizosaccharomyces pombe (Affymetrix and Agilent), or Candida albicans (Washington University, St. Louis). Proteomic studies are also hampered as they rely on annotated genome sequences for efficient performance. Alternatively, research groups performed transcriptional profiling by either heterologous hybridization to commercial S. cerevisiae arrays (e.g. for P. pastoris, Sauer, 2004; and for K. lactis, Rosende, 2008), or by designing custom microarrays. As an example, P. pastoris microarrays have been developed by our group, and are available for research applications (Graf, 2008). While the first approach can only capture genes that are in common with S. cerevisiae [therefore excluding species-specific genes such as the assimilation pathways for methanol (P. pastoris and Hansenula polymorpha), hydrocarbons (Yarrowia lipolytica), or xylose (Pichia stipitis)], the latter often made the custom-made arrays unavailable for other groups, thereby limiting research activities in the field. Consistently, proteomics were mainly performed for pathogenic species (reviewed by Josic & Kovac, 2008).
Another drawback in the fungal kingdom is the high genetic diversity between the individual species, even within the phylum Ascomycota. The average sequence identity of orthologous proteins among the hemiascomycota is 50–60%, which is less than the c. 70% identity between man and fish, not to speak of 94% identity between man and mouse (Dujon, 2006). Accordingly, the DNA sequence identity among rodents is much higher, making heterologous omics between the hamster-derived Chinese hamster ovarian cells (CHO) or baby hamster kidney (BHK) cells and mouse or rat more feasible than among yeasts.
Filamentous fungi
A recent publication summarized ‘The first 50 microarray studies in filamentous fungi’, starting with an incomplete microarray for Trichoderma reesei in 2002 (Breakspear & Momany, 2007) and stated that for filamentous fungi, so far no engineering based on omics existed. However, in 2008 the first reports of proteome and transcriptome studies resulting in clear engineering strategies were published (Table 2): 132 protease genes were monitored during the production of human lysozyme in Aspergillus oryzae on a microarray and compared with degradation conditions, and upregulated protease cluster were identified. Out of these disruption targets three genes were already known to improve heterologous protein production, but the knock-down of one novel protease improved secreted yields of human lysozyme by 22% (Kimura, 2008). Wang (2008) identified four protease genes of Aspergillus niger with genomic methods, which, upon disruption, led to increased protein secretion up to 40%. Another study determined the effect of enzyme overproduction in three different strains of A. niger, and extracted two engineering targets out of the upregulated genes and proteins involved in protein folding and the endoplasmic reticulum (ER)-associated protein degradation (ERAD) pathway. Combined engineering by knock-out of the ERAD factor doaA and overexpression of the oligosaccharyltransferase sttC led to improved production of a heterologous protein (Jacobs, 2009).
However, it should be noted that a number of genome-wide studies have revealed important aspects concerning protein production and its limitation in fungal expression systems. While not being applied directly, they have contributed significantly to the understanding of the protein production process, and led to strain engineering later on. The transcriptomic changes upon protein overexpression have been described for Aspergillus nidulans (Sims, 2005), T. reesei (Arvas, 2006) and A. niger (Guillemette, 2007), highlighting the impact of unfolded protein response (UPR) on protein folding, glycosylation, vesicle transport and ERAD, and identifying significant differences of UPR regulation between S. cerevisiae and filamentous fungi.
If we look beyond fungi, examples for systems biotechnological approaches in the field of recombinant protein production become more prevalent. Since the late 1990s, transcriptomics and proteomics were applied to bacterial and mammalian cultures used for heterologous protein production in order to elucidate cell physiology. Although numerous studies exist that describe the physiological behaviour of cells to certain stresses – data also available for S. cerevisiae– activity beyond pure description is concentrated to a limited number of research groups. Recent reviews by these groups highlight that it is crucial to use state-of-the-art omics tools for physiological understanding and gaining insights into the host, as only detailed understanding of host cell physiology makes subsequent metabolic or cell engineering possible.
Mammalian cells
In mammalian cell culture most proteomic and transcriptomic analyses were performed to address problems or phenomena that have been observed previously on a ‘macroscopic’ level (e.g. metabolic shift, fed batch cultivation, apoptosis and stress conditions brought up by elevated osmolarity, sodium butyrate and low temperature). Since Korke (2002) predicted the implementation of genomics and proteomics in cell culture engineering – for example for the selection of production cell lines (identification of gene regulation leading to adaptation to serum-free growth, or to adaptation to suspension growth) – and for bioprocess engineering both methodology and answers have evolved. Several excellent reviews summarizing these recent advances have been published during the last years (Griffin, 2007; Gupta & Lee, 2007; Kuystermans, 2007; Jaluria, 2008). A list of papers dealing with genome-wide analysis of different mammalian cell lines (mainly murine myeloma cells NS0 and CHO) can be found in Kuystermans (2007). In agreement with Gupta & Lee (2007), who pinpoint that a large number of omics approaches only generate lists of genes without direct application, we confirm that only four out of the 21 cited studies have resulted in an actual strategy for cell line engineering (Table 2).
Dinnis & James (2005) asked if one should learn ‘lessons from nature’ for engineering of antibody secreting mammalian cells. At least two lessons have been learned: induction of the UPR in order to reflect B-cell development (van Anken, 2003) leads to increased secretion of several recombinant proteins (reviewed by Dinnis & James, 2005; Khan & Schroder, 2008). Another lesson that has been learned was the overexpression of anti-apoptotic genes, and knock-down of pro-apoptotic genes identified by microarray analysis of CHO cells, leading to prolonged cell viability and consequently up to 2.5-fold higher titres of interferon γ (Wong, 2006).
Engineering mammalian cell culture based on genome-scale technologies was mainly applied to improve cell metabolism and growth (Griffin, 2007), upstream cell culture conditions (e.g. temperature, hyperosmotic pressure and impact of small chemical compounds), downstream product quality (mainly assessed by proteomics), and cell culture media requirements (Gupta & Lee, 2007). As an example, the transcriptional analysis and proteomics did not stop at the identification of the responsible genes for the cholesterol dependence of NS0 cells. Subsequent engineering of the identified genes allowed cholesterol-independent cell growth (Seth, 2006). Cross-species microarrays of high producer clones of EPO-Fc producing CHO lead to the identification of three ER stress marker genes correlated to insufficient resistance to shear stress in the early stage of clone selection before the respective phenotype could be observed (Trummer, 2008).
Proteomic and transcriptional profiling of high- and low-productivity cell lines, or cells cultivated under conditions that lead to high specific productivity (qP) (e.g. treatment with sodium butyrate and low-temperature cultivation) were carried out to discover the target genes leading to the super-secreting cells. Common features correlated to high productivity were the upregulation of secretory pathway proteins (especially chaperones and foldases) and cytoskeletal proteins in high-producing cell lines, as well as higher abundance of proteins belonging to the functional groups redox balance and vesicular transport. Additionally, decreased growth rate-related genes/proteins and decreased levels of stress genes were reported to occur in concordance with higher qP (Kuystermans, 2007; Seth, 2007). While there were speculations that high-producing cell lines are likely to have a higher vesicle traffic and membrane recycling activity (Yee, 2008), other attempts to identify correlations between single genes and improved secretory capacity failed. When trying to integrate all available genome-scale information of high-producing mammalian cell lines to find the genetic events leading to the super-secreting cells, it had to be concluded that there is no direct relation between a distinct set of genes and a trait, that there are no ‘hyperproductivity master genes’ (Seth, 2007). On the contrary, multiple contributing pathways, even alternative pathways may lead to improved qP. Therefore the authors highlight the importance of data analysis approaches going beyond the identification of differentially expressed genes such as pattern discovery, pathway and network analysis in order to grasp the complexity of the gene–trait relationship (Seth, 2007).
Bacteria
A general overview about whole systems level metabolic engineering in bacteria based on omics, including potential applications, was given by Park (2005) and Gupta & Lee (2007). One common feature in bacteria, for example Escherichia coli, is that protein overproduction usually leads to a (severe) decrease in specific growth rate due to a shortage of energy and precursors.
Very early proteomic work in E. coli and Bacillus subtilis overproducing a heterologous protein accumulating in the cytoplasm as inclusion bodies showed that both species react to the recombinant protein with increased levels of heat shock proteins and chaperones, whereas no clear picture regarding the regulation of ribosomal proteins emerged, as higher abundance in B. subtilis, and decreased levels in E. coli were observed (Jürgen, 2000, 2001). These studies can be seen as initiating a comprehensive understanding of the cellular responses to protein overproduction in bacteria, and although no direct engineering benefits were achieved, they gave rise to improvement of production strains (reviewed by Chou, 2007).
Since then various studies investigated the response of bacteria to several different proteins, but hardly any new hypotheses or applications evolved out of these studies. Some rare exceptions to this include the overexpression of ribosomal genes downregulated during insulin-like growth factor 1 expression in high cell density cultivation of E. coli leading to enhanced productivity and the engineering of small heat shock proteins IbpAB identified in inclusion bodies during overexpression of recombinant proteins in E. coli (all summarized in Park, 2005). By proteome profiling Aldor (2005) identified the phage shock protein PspA to be coregulated with heterologous protein expression, and improved the yield by controlled coexpression of PspA. However, most studies are conducted on a case-by-case basis, and are not leading to improved production platforms. Alternatively, genome-wide analysis of cellular reactions to protein production may allow for the identification of marker genes that signal cellular stress as a response to protein overexpression. Their monitoring during protein production processes should allow to react on the bioprocess level before the stressful conditions, and consequently reduced cell growth and reduced viability will occur (Dürrschmid, 2008; Nemecek, 2008).
Interestingly, genome-wide analyses were also performed to analyse the behaviour of mutant strains with superior production characteristics. As an example, the proteome of a Bacillus megaterium chemical mutant exhibiting higher production levels of recombinant intracellular dextranesucrase and better cultivation behaviour, showed higher abundance of proteins related to protein synthesis and protein translocation (Wang, 2006). The observation of reduced levels of tRNA synthetases both on the proteomic and the mRNA level of an E. coli mutant secreting four times more α-haemolysin (HylA) in comparison with its parental strain led to an alternative metabolic engineering strategy, namely to use rare codons to slow down translation, which improved HylA secretion eight times in the parental strain (Lee & Lee, 2005).
In a different application of omics technologies, proteome analysis of E. coli in response to oleic acid was used to select oleic acid-inducible promoters for recombinant protein production. The use of the aldehyde dehydrogenase aldA promoter increased green fluorescent protein fluorescence intensity 30-fold compared with the IPTG-inducible tac promoter while applying the cheaper inducer, oleic acid (Han, 2008). On the other hand, a screen of the extracellular proteome of E. coli identified naturally secreted proteins as fusion partner for recombinant proteins in order to stimulate secretion. Out of 12 tested low-molecular-weight fusion partners, OsmY proved to be the best secretion partner resulting in high-level excretion of three model proteins into the culture supernatant of E. coli (Qian, 2008).
Alternatively, metabolic flux calculations can be carried out with the aim to identify bottlenecks in the fermentation that may need to be eliminated by genetic engineering. A recent study investigated the influence of two different carbon sources (glucose and pyruvate) on metabolic fluxes and productivity in B. megaterium, and concluded that pyruvate improves recombinant protein production 17-fold as less protease secretion and enhanced energy and reduction equivalent metabolism occurred. Additionally, the authors state that the overproduction of the recombinant protein increases the flux through the TCA and glycolysis, and reduces the flux through gluconeogenesis and the pentose phosphate pathway (Fürch, 2007).
New (post) genomic approaches to systems biotechnology
Systems biology as well as application-oriented systems biotechnology depend essentially on omics methods. A critical overview on current developments in this area and the potentials and pitfalls of current and upcoming methods is provided in the following, and summarized in Table 3.
Critical summary of omics methods for systems biotechnology
| Field | Methods | Advantages and disadvantages | ||
| Genomics | NGS | Fast and cheap method for whole-genome (re)sequencing without cloning bias Advantageous for mutation and subsequent strain analysis for inverse metabolic engineering purposes | ||
| Transcriptomics | Expression microarrays | Whole genome transcriptomics, cheap solution for in-house pipeline Susceptible to noise and bias | ||
| Ref-Seq | Better correlation to qPCR results, large dynamic range limited only by sequencing depth Little background noise Can be used to detect splicing variants and 5′- and 3′-UTR boundaries Quantification is feasible even for mRNAs expressed at low levels Loss of strand-specific information | |||
| Proteomics | DIGE (gel-based systems) | Large number of different proteins over a large mass range can be detected Information about physicochemical properties Expensive and biased towards high-abundant proteins Membrane-bound and hydrophobic as well as small proteins cause problems | ||
| MS | Mass and structure information of proteins Amino acid composition Detection of post-translational modifications | |||
| Quantitative MS | Labelling (in vivo– SILAC, in vitro– ICAT, iTRAQ): increases the dynamic range of the analysis, but more expensive and detected proteins depend on the labelling method; in vivo labelling is not suitable for industrial processes Label-free quantification: quantification of a large number of proteins to characterize cells in different states; but less accurate and problematic to identify low-abundant proteins | |||
| Protein Microarrays | Can only detect selected proteins due to lack of highly specific capture reagents and a lack in sensitivity Difficulties in retaining protein functionality | |||
| Interactomics | ChIP-chip | Regulatory DNA–protein binding interactions Chromatin packaging | ||
| ChIP-Seq | Better resolution, less input material needed than ChIP-chip Usable for organisms without available genomic sequence Quantification is possible | |||
| Metabolomics | Metabolic modelling | Understanding regulatory pathways Identifying key players Simulation of system- wide reactions (either through logical networks or flux analysis) before biotechnological engineering is possible Creation of metabolic/signaling networks is complex and time consuming | ||
| Field | Methods | Advantages and disadvantages | ||
| Genomics | NGS | Fast and cheap method for whole-genome (re)sequencing without cloning bias Advantageous for mutation and subsequent strain analysis for inverse metabolic engineering purposes | ||
| Transcriptomics | Expression microarrays | Whole genome transcriptomics, cheap solution for in-house pipeline Susceptible to noise and bias | ||
| Ref-Seq | Better correlation to qPCR results, large dynamic range limited only by sequencing depth Little background noise Can be used to detect splicing variants and 5′- and 3′-UTR boundaries Quantification is feasible even for mRNAs expressed at low levels Loss of strand-specific information | |||
| Proteomics | DIGE (gel-based systems) | Large number of different proteins over a large mass range can be detected Information about physicochemical properties Expensive and biased towards high-abundant proteins Membrane-bound and hydrophobic as well as small proteins cause problems | ||
| MS | Mass and structure information of proteins Amino acid composition Detection of post-translational modifications | |||
| Quantitative MS | Labelling (in vivo– SILAC, in vitro– ICAT, iTRAQ): increases the dynamic range of the analysis, but more expensive and detected proteins depend on the labelling method; in vivo labelling is not suitable for industrial processes Label-free quantification: quantification of a large number of proteins to characterize cells in different states; but less accurate and problematic to identify low-abundant proteins | |||
| Protein Microarrays | Can only detect selected proteins due to lack of highly specific capture reagents and a lack in sensitivity Difficulties in retaining protein functionality | |||
| Interactomics | ChIP-chip | Regulatory DNA–protein binding interactions Chromatin packaging | ||
| ChIP-Seq | Better resolution, less input material needed than ChIP-chip Usable for organisms without available genomic sequence Quantification is possible | |||
| Metabolomics | Metabolic modelling | Understanding regulatory pathways Identifying key players Simulation of system- wide reactions (either through logical networks or flux analysis) before biotechnological engineering is possible Creation of metabolic/signaling networks is complex and time consuming | ||
Critical summary of omics methods for systems biotechnology
| Field | Methods | Advantages and disadvantages | ||
| Genomics | NGS | Fast and cheap method for whole-genome (re)sequencing without cloning bias Advantageous for mutation and subsequent strain analysis for inverse metabolic engineering purposes | ||
| Transcriptomics | Expression microarrays | Whole genome transcriptomics, cheap solution for in-house pipeline Susceptible to noise and bias | ||
| Ref-Seq | Better correlation to qPCR results, large dynamic range limited only by sequencing depth Little background noise Can be used to detect splicing variants and 5′- and 3′-UTR boundaries Quantification is feasible even for mRNAs expressed at low levels Loss of strand-specific information | |||
| Proteomics | DIGE (gel-based systems) | Large number of different proteins over a large mass range can be detected Information about physicochemical properties Expensive and biased towards high-abundant proteins Membrane-bound and hydrophobic as well as small proteins cause problems | ||
| MS | Mass and structure information of proteins Amino acid composition Detection of post-translational modifications | |||
| Quantitative MS | Labelling (in vivo– SILAC, in vitro– ICAT, iTRAQ): increases the dynamic range of the analysis, but more expensive and detected proteins depend on the labelling method; in vivo labelling is not suitable for industrial processes Label-free quantification: quantification of a large number of proteins to characterize cells in different states; but less accurate and problematic to identify low-abundant proteins | |||
| Protein Microarrays | Can only detect selected proteins due to lack of highly specific capture reagents and a lack in sensitivity Difficulties in retaining protein functionality | |||
| Interactomics | ChIP-chip | Regulatory DNA–protein binding interactions Chromatin packaging | ||
| ChIP-Seq | Better resolution, less input material needed than ChIP-chip Usable for organisms without available genomic sequence Quantification is possible | |||
| Metabolomics | Metabolic modelling | Understanding regulatory pathways Identifying key players Simulation of system- wide reactions (either through logical networks or flux analysis) before biotechnological engineering is possible Creation of metabolic/signaling networks is complex and time consuming | ||
| Field | Methods | Advantages and disadvantages | ||
| Genomics | NGS | Fast and cheap method for whole-genome (re)sequencing without cloning bias Advantageous for mutation and subsequent strain analysis for inverse metabolic engineering purposes | ||
| Transcriptomics | Expression microarrays | Whole genome transcriptomics, cheap solution for in-house pipeline Susceptible to noise and bias | ||
| Ref-Seq | Better correlation to qPCR results, large dynamic range limited only by sequencing depth Little background noise Can be used to detect splicing variants and 5′- and 3′-UTR boundaries Quantification is feasible even for mRNAs expressed at low levels Loss of strand-specific information | |||
| Proteomics | DIGE (gel-based systems) | Large number of different proteins over a large mass range can be detected Information about physicochemical properties Expensive and biased towards high-abundant proteins Membrane-bound and hydrophobic as well as small proteins cause problems | ||
| MS | Mass and structure information of proteins Amino acid composition Detection of post-translational modifications | |||
| Quantitative MS | Labelling (in vivo– SILAC, in vitro– ICAT, iTRAQ): increases the dynamic range of the analysis, but more expensive and detected proteins depend on the labelling method; in vivo labelling is not suitable for industrial processes Label-free quantification: quantification of a large number of proteins to characterize cells in different states; but less accurate and problematic to identify low-abundant proteins | |||
| Protein Microarrays | Can only detect selected proteins due to lack of highly specific capture reagents and a lack in sensitivity Difficulties in retaining protein functionality | |||
| Interactomics | ChIP-chip | Regulatory DNA–protein binding interactions Chromatin packaging | ||
| ChIP-Seq | Better resolution, less input material needed than ChIP-chip Usable for organisms without available genomic sequence Quantification is possible | |||
| Metabolomics | Metabolic modelling | Understanding regulatory pathways Identifying key players Simulation of system- wide reactions (either through logical networks or flux analysis) before biotechnological engineering is possible Creation of metabolic/signaling networks is complex and time consuming | ||
Genomes
As already discussed in the previous section systems biotechnology has focused on certain organisms simply because their genome was sequenced and at least partly annotated. This is understandable because many omics methods can only be utilized to their full extent if the genome sequence is accessible and information about the positions of functional elements in the DNA is available. Furthermore, though the genome of S. cerevisiae has been very well studied, it is not a typical example for many yeast species that are used for protein production (Blank, 2005). Therefore, it is vital for systems biotechnology to create reference genomes with high-quality annotation of yeast species used in protein production. Sequencing technologies have made tremendous progress in the last few years, rendering the technology significantly cheaper, faster and more flexible than the traditional Sanger method, making it feasible for small scale studies with limited resources. With a reference genome at hand resequencing becomes an integral part of the workflow in systems biotechnology as shown in Fig. 3, thus expanding the systems biotechnology cycle. First studies applying the technology to selected mutants to understand the genetic background of their improved phenotype are already available for ethanol-producing P. stipitis (Smith, 2008).
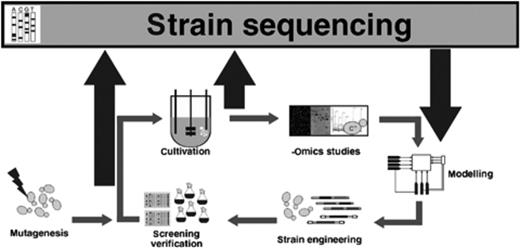
A second genomics based systems biotechnology circle. NGS methods enable the resequencing of selected mutants to superimpose genotype on phenotype. Thus, evolutionary engineering and inverse metabolic engineering will gain enormous new potential.
Next generation sequencing (NGS)
Though NGS has a high potential of revolutionizing genetics, it comes with a set of pitfalls. All NGS methods create much shorter sequence reads (35–400 bp) than the Sanger method (c. 750–900 bp). This is especially a problem for de novo sequencing because even short repeats will make an assembly impossible, resulting in a high number of contigs. A comprehensive summary of the NGS technologies that are currently available was published by Mardis (2008). Third-generation sequencing (TGS) or also called next-next generation sequencing methods aim to further reduce the cost and run time of sequencing while improving the ease of handling the method. Additionally, most of these new technologies promise to have much longer reads than the NGS methods and thereby eliminating the problems related to short read length. Variants of currently pursued TGS technologies could be commercially available within the next 5 years (Gupta, 2009).
After the first excitement of the 1990s about the possibilities of sequencing and the completion of the human genome project in 2003, it was believed that the postgenomic era had begun. Now, a few years later, the picture looks somewhat different. It seems that with the emergence of NGS, the chapter of genomics has to be reopened again. At the moment, there are 873 completely sequenced genomes, of which about 83% are bacterial species having a rather small genome, and 4135 ongoing genome projects with a much lower proportion (50%) of bacterial species (http://www.genomesonline.org/gold.cgi). The rapidly increasing amount of available genomic data poses a challenging problem for data storage, management and interpretation, shifting the bottleneck towards bioinformatics, annotation and analysis tools.
Transcriptomics
Apart from de novo sequencing, NGS and TGS methods can be applied to many other questions that are relevant for the optimization of protein production, for example copy number variation, transcription factor binding, noncoding RNAs as well as expression profiles. Up to now DNA microarrays were the technologies of choice in the field of transcriptomics. For organisms for which microarrays are not available but that have an accessible genome sequence, bioinformatics tools (gene prediction and oligo design) make it possible to generate comprehensive expression data at a relatively low cost, as recently shown for P. pastoris (Graf, 2008). Microarray experiments generally suffer from the existence of nonbiological variation, high background noise and only limited comparability due to a multitude of possible data-processing methods (Kawasaki, 2006). Sources for nonbiological variability and possible computational solutions have been extensively discussed in the literature (e.g. Draghici, 2006; Johnson, 2008). NGS solves many of the technical problems that microarrays suffer from but is still more expensive and many research groups and companies have an established microarray-processing pipeline in place. Also, some companies that offer NGS-sequencing machines offer microarrays as well, which indicates that a complementary use of both technologies is the most probable future.
Proteomics
Changes in the behaviour of a production system are often affected by post-translational modifications of proteins and therefore not visible on a transcriptome level. Besides there is no quantitative correlation between transcript expression levels and the amount of protein in the cell (Hartmann, 2008). Analysing the genome and transcriptome alone is therefore not sufficient to understand or predict regulatory mechanism of cells or organisms. Unfortunately, though proteomics provides valuable insights for systems biotechnology, studies of changes during protein production are largely still missing (Josic & Kovac, 2008). The classical technique of proteome research, two-dimensional (2D) gel electrophoresis, suffers from a bias towards specific protein classes and towards highly abundant proteins (Bro & Nielsen, 2004) on the experimental level. On the data-processing level analysis of 2D gel images comprises many of the problems of microarray analysis, especially the difficulty to compare results due to differences between platforms and data-processing methods (Elrick, 2006). In recent years, proteomics moved from a local (analysing a limited selection of proteins) to a global (analysing the whole proteome) technology. Quantitative mass spectroscopy has become the method of choice, and coupled with more sensitive labelling methods it facilitates high throughput proteome analysis (Elrick, 2006). Stable isotope labelling by amino acids in cell culture (SILAC) is an in vivo labelling technique that requires the cells to be cultivated in media-containing labelled amino acids, thereby rendering it not feasible for large-scale production analyses. In vitro labelling techniques are more promising, although they still have shortcomings. In isotope-coded affinity tags (ICAT) cysteine residues are tagged. Because these residues are rare, it simplifies the peptide mixtures but proteins that do not contain cystein cannot be measured, and, furthermore, the small number of peptides for each protein compromises measurement reliability. Another in vitro labelling method is isobaric tags for relative and absolute quantification (iTRAQ). Here the N-terminus and sidechain amines are tagged with at least four different masses. Because these amines are more frequent, the cysteine-based restriction of ICAT is removed (Bachi & Bonaldi, 2008). Because all approaches involving isotopes are cost-intensive, label-free quantification methods using spectrum counts, integrated ion intensities or spectral feature analysis are sometimes preferable. The drawbacks of label-free methods include increased computational complexity due to lower accuracy and reproducibility of the data and the inability to quantify low-abundance proteins (Nesvizhskii, 2007). With the move towards high throughput, it is essential to develop and use the proper bioinformatics tools to efficiently process and statistically validate the generated data.
Metabolomics and metabolic modelling
Metabolomics and fluxomics go a step further and use information gained from the other omics to build a model of certain processes of the cell or ideally of the whole cell. Whereas metabolomics focuses on the metabolites involved, fluxomics predicts flux distributions within the cell based on measured rates of metabolites and their mass balance (Kim, 2008). Several types of metabolic networks exist. Stoichiometric models and dynamic or kinetic models are the more traditional approach for which comprehensive knowledge about the players and their relationship is necessary. These models consist of a relatively small number of reactions or elements and their quality depends to a large extent on the quality of experimental data (Steuer, 2007). The advantage is of course that, if a high-quality model is achieved, reasonable predictions about phenotypic behaviour can be made. Early metabolic networks were limited to a few pathways of the core metabolism, but the availability of genome sequences and extensive omics data made it possible to fully describe the core metabolism and move to new areas such as signalling (Arga, 2007) or lipid metabolism (Nookaew, 2008). Protein expression and secretion networks are still lacking. Some of the reasons why this topic was avoided by scientists for so long are listed in Table 1. Despite these pitfalls, it can be anticipated that model development of the protein production pathways will significantly contribute to a better quantitative understanding of the contributing reactions and their relevance.
Topological networks on the other hand consist of many nodes that represent genes or proteins and edges representing the connection between those nodes. Such models can be much larger than stoichiometric or kinetic models and can make use of high throughput data but are only static descriptions and contain no information about the type, time or place of interaction between two nodes. At least the type of interaction can be modelled using control logic, described by Schlitt & Brazma (2005). With a sequenced and annotated genome at hand, a topological or control logic network can be computed for all known cellular functions. The challenge now is to combine the different approaches into a predictive dynamic model of the whole cell. The first such approach was taken for A. niger (Andersen, 2008), while Herrgård (2008) took the first step towards such a model for S. cerevisiae by combining two existing metabolic models (at the control logic state) into a consensus yeast metabolic network and implementing it in systems biology markup language (SBML), which is a widely used data format for metabolic networks.
With the pace at which omics methods develop and the amount of data they produce, it is important to keep in mind that each of the omics fields only shows us an isolated part of the picture. To improve our understanding of the function of organisms, it is essential to be able to give meaning to the data representing concentrations of genes, proteins and metabolites. Therefore, it will become even more important in the future to merge information from different omics sources into a coherent picture. More effort has to be put in developing methods that can integrate and validate these data as well as help managing the fast-increasing flood of information. This in mind, several standards have been developed to avoid a confusing mess of data sources, treatment and analysis variants (Table 4). While these standards define a minimal frame on experimental and computational quality and data deposition, they cannot cope with systematic differences between different omics platforms. These systematic differences call for cautiousness in data comparison between omics platforms and data-processing methods. It should be emphasized that the aim of standardization is not to define data quality, but rather the information on data assessment, and to guarantee that raw data are made available to the scientific community.
Standards for the publication of omics data
| Short name | Designation | References or web addresses |
| MIAME | Minimum information about microarray experiments | http://www.mged.org/Workgroups/MIAME/miame.html |
| MIAPE | The minimum information about a proteomics experiment | http://www.psidev.info/index.php?q=node/91 |
| MIRIAM | Minimum information requested in the annotation of biochemical models | http://www.ebi.ac.uk/compneur-srv/miriam/, proposed by Le Novère (2005) |
| MIAMET | Minimum information on a metabolomics experiment | Proposed by Bino (2004) |
| Short name | Designation | References or web addresses |
| MIAME | Minimum information about microarray experiments | http://www.mged.org/Workgroups/MIAME/miame.html |
| MIAPE | The minimum information about a proteomics experiment | http://www.psidev.info/index.php?q=node/91 |
| MIRIAM | Minimum information requested in the annotation of biochemical models | http://www.ebi.ac.uk/compneur-srv/miriam/, proposed by Le Novère (2005) |
| MIAMET | Minimum information on a metabolomics experiment | Proposed by Bino (2004) |
Standards for the publication of omics data
| Short name | Designation | References or web addresses |
| MIAME | Minimum information about microarray experiments | http://www.mged.org/Workgroups/MIAME/miame.html |
| MIAPE | The minimum information about a proteomics experiment | http://www.psidev.info/index.php?q=node/91 |
| MIRIAM | Minimum information requested in the annotation of biochemical models | http://www.ebi.ac.uk/compneur-srv/miriam/, proposed by Le Novère (2005) |
| MIAMET | Minimum information on a metabolomics experiment | Proposed by Bino (2004) |
| Short name | Designation | References or web addresses |
| MIAME | Minimum information about microarray experiments | http://www.mged.org/Workgroups/MIAME/miame.html |
| MIAPE | The minimum information about a proteomics experiment | http://www.psidev.info/index.php?q=node/91 |
| MIRIAM | Minimum information requested in the annotation of biochemical models | http://www.ebi.ac.uk/compneur-srv/miriam/, proposed by Le Novère (2005) |
| MIAMET | Minimum information on a metabolomics experiment | Proposed by Bino (2004) |
Conclusions
Current status of systems biotechnology for protein production
Systems biotechnology has proven its value for strain design and optimization. System-wide approaches to complex cellular processes such as protein production are still in their infancy. Interestingly, more progress in this field has been made for bacteria and mammalian cells than for yeasts. This may be due to a lack of genomic information and postgenomic tools for industrially relevant yeast species. The revival of genomics through NGS methods is about to close this gap, and a critical review of the state of research with bacterial and cell culture host systems provides guidance as to where to direct research with yeasts in this field.
Lessons to learn from non-yeast expression systems
At present, most limitations in protein production in yeasts are attributed to bottlenecks during folding and secretion. Thus, engineering of yeast protein factories mainly means knowledge-based engineering of chaperones and ER resident folding catalysts. While these approaches were verified by transcriptomic profiling of yeasts and other fungal production hosts recently, they may not convey the full picture. As can be seen in non-yeast expression systems, the application of integrated genomic approaches allows looking beyond the borders of the secretory compartments during the production of recombinant proteins. While engineering strategies leading to higher viability and higher stress resistance in mammalian cells may not be directly applied to yeast systems, they reveal the potential that apparently unconnected cellular processes can be manipulated in order to increase protein yields/productivity. Bacterial studies revealed a shortage of ribosomes, energy and precursors during recombinant protein production, a possibility yet underestimated for fungal hosts. The availability of improved metabolic models – as they exist for bacteria – for A. niger and S. cerevisiae, and their applicability for related fungal species, may allow a considerable progress regarding the behaviour of the core metabolism and energy supply during protein overexpression. Alternatively, production processes can be monitored by methods of metabolic flux analysis and controlled to improve protein yields.
The importance of systems level screening during clone selection in mammalian cells may also be converted to fungal production hosts, as soon as the respective genetic/molecular traits leading to high secretory capacity are identified. Once again, the importance of thorough and comparative data analysis should be stressed in this respect, as the pathways identified upon protein overproduction in yeasts and filamentous fungi are very similar to those regulated in mammalian cells.
With the advent of cheap and fast sequencing technologies, and consequently higher coverage of biotechnologically relevant fungal species, resequencing of improved mutant strains and subsequent inverse metabolic engineering also become feasible. Additionally, this information may contribute to the big search for the ‘holy grail’ of protein production – the ‘hyperproductivity master genes’– or at least lead to a better understanding of the cellular pathways influencing productivity.
Finally, the impact of systems biotechnology on the improvement of bioprocess performance, for example by the identification of novel stress markers such as those shown in E. coli, better strain performance (mammalian cells) or prevention of proteolytic degradation by disruption of cellular proteases as reported for filamentous fungi and bacterial systems should be highlighted. All these approaches can be readily transferred to fungal production processes by applying the respective systems biotechnological tools.
In contrast to mammalian and bacterial protein production, application of systems level approaches for the targeted engineering of yeasts and other fungal production hosts is still at an early stage. However, expectations are high that the recent advances emerging in the fungal field will just be the beginning of the ‘systems biotechnological’ age for improved protein production strains.
Outlook
A major focus of future work should be the quantitative understanding of molecular principles behind protein synthesis, modification and secretion, derived from basic production strains as well as mutants and rationally engineered strains.
NGS methods provide the tool to rationalize inverse metabolic engineering approaches so that they can be implemented in future into rational system-wide modelling and optimization strategies.
Acknowledgements
Research on heterologous protein production in yeasts in our laboratory is supported by the Austrian Science Fund (project no. I37-B03), the European Science Foundation (programme EuroSCOPE), the Austrian Research Promotion Agency (programme FHplus), Polymun Scientific GmbH and Boehringer Ingelheim Austria GmbH.
References
Author notes
Editor: Teun Boekhout

{kind=link}
{kind=link}
{kind=link}